031186508999
Xyb@xuekang.com

【通用名称】 艾伏尼布片
【商品名称】 拓舒沃
【英文名称】 Ivosidenib Tablets
【汉语拼音】 Ai Fu Ni Bu Pian
警示语:
分化综合征
本品治疗的患者可能发生分化综合征,如果不进行治疗,可导致死亡。当考虑发生分化综合征时,应给予患者皮质类固醇治疗,并监测血流动力学,直到患者症状消退(参见【注意事项】和【不良反应】)。
成份:
本品主要成份为艾伏尼布。
化学名称:(2 S)-N-[ (1 S)-1-(2-氯苯基)-2-[(3,3-二氟环丁基)氨基]-2-氧代乙基]-1-(4-氰基吡啶-2-基)-N–(5-氟吡啶-3-基)-5-氧代吡咯烷-2-甲酰胺
化学结构式:
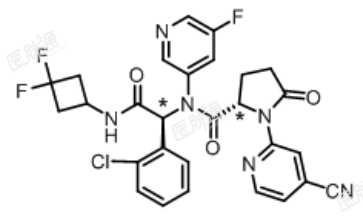
分子式:C28H22ClF3N6O3
分子量:583.0
辅料:醋酸羟丙甲纤维素琥珀酸酯、微晶纤维素、交联羧甲基纤维素钠、十二烷基硫酸钠、胶态二氧化硅、硬脂酸镁、水溶性薄膜包衣
所属类别:
化药及生物制品 >> 抗肿瘤药和免疫机能调节药 >> 抗肿瘤药 >> 其它抗肿瘤药物
化药及生物制品 >> 其他 >> 其他
适应症:
本品适用于采用经充分验证的检测方法诊断为携带易感异柠檬酸脱氢酶-1(IDH1)突变的复发性或难治性急性髓系白血病(AML)成人患者。
IDH1 突变的检测要求详见【用法用量】。
本品为临床急需品种,基于境外一项单臂临床试验的缓解率以及中国受试者药代动力学数据获得附条件批准上市,治疗中国患者的有效性和安全性尚待上市后进一步确证。
规格:
0.25 g
用法用量:
患者选择
在使用本品治疗复发性或难治性急性髓系白血病(AML)成人患者之前,必须确定患者骨髓或外周血中具有异柠檬酸脱氢酶-1(IDH1)突变。应采用充分验证过的检测方法确定患者的 IDH1 突变状态。经医院或实验室的 IDH1 突变检测结果判断为携带 IDH1 突变的患者能接受本品治疗,并且应在基石药业(苏州)有限公司指定的实验室使用研究性伴随诊断检测方法对患者的 IDH1突变状态进行再次检测,检测结果证实患者确伴有 IDH1 突变可继续用药。
推荐剂量
本品推荐剂量为 500 mg,每日一次口服,直至疾病进展或出现不可接受的毒性。对未出现疾病进展或不可接受毒性的患者,需至少接受 6 个月治疗,以充分观察临床反应。
本品可空腹或餐后口服。服药时,不要进食高脂肪餐,以免导致血药浓度增加(参见【药代动力学】)。
不要掰开或碾碎本品服用。
本品每天应在固定时间服用。
如果服药后出现呕吐,不需补服;按照预定时间进行下一次服药。
如果漏服或未在既定时间服药,应尽快补服;但如果距下一次预定服药时间小于 12 小时,则无需补服,第二天恢复原计划时间服药即可。12 小时内不得服药 2 次。
针对毒性的监测
首次给药前、治疗的第一个月至少每周一次、治疗的第二个月每两周一次,此后治疗期间每月一次检查血细胞计数和血生化。在治疗的第一个月,每周一次监测血肌酸磷酸激酶。在治疗的前三周至少每周一次心电图(ECG)检查,此后,在治疗期间至少每月检查一次 ECG。有任何异常发现均需及时处理(参见【不良反应】)。
针对毒性的剂量调整
应根据毒性反应暂停服药或降低剂量。关于剂量调整方案见表 1。
表 1. 本品推荐的剂量调整方案
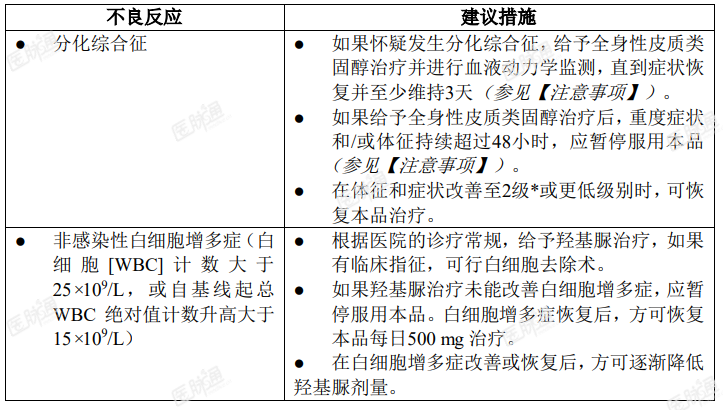
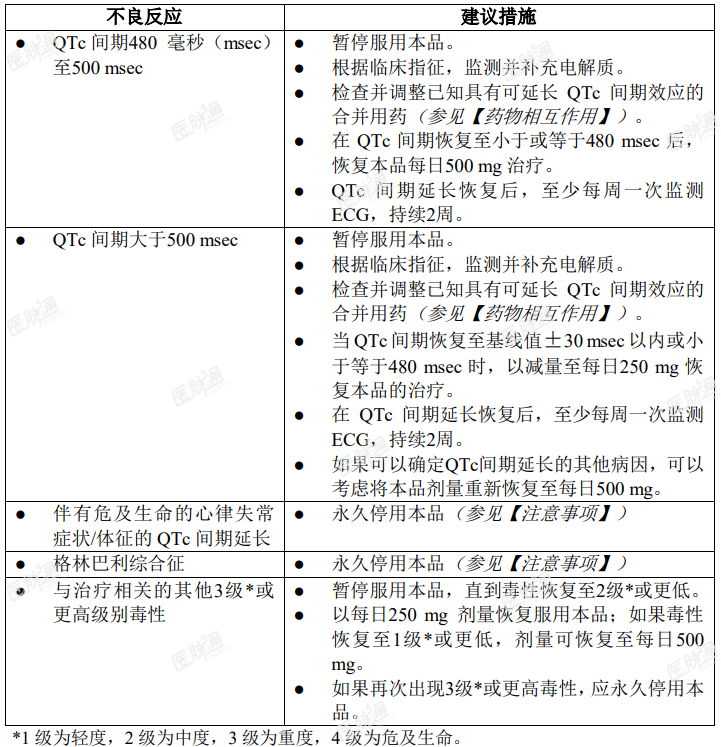
与强 CYP3A4 抑制剂合并给药的剂量调整
如果必须与强 CYP3A4 抑制剂合并给药,应将本品剂量降低至 250 mg,每 日一次。在强 CYP3A4 抑制剂治疗停止后至少 5 个半衰期,可将本品恢复至推 荐剂量 500 mg,每日一次。
特殊人群
肝功能不全
对于轻度或中度(Child-Pugh A 或 B)肝功能损害的患者,无需调整起始剂量。 艾伏尼布片在重度肝功能损害(Child-Pugh C)患者中的药代动力学和安全性尚不明确。对于预先存在重度肝功能损害患者,在开始使用艾伏尼布片治疗之前,应考虑其风险和潜在获益。
肾功能不全
对于轻度或中度肾功能损害(eGFR ≥ 30 mL/min/1.73m2,MDRD)的患者, 无需调整起始剂量。 艾伏尼布在患有重度肾功能损害(eGFR < 30 mL/min/1.73m2,MDRD)或需要透析的肾功能损害患者中的药代动力学和安全性尚不明确。对于预先存在重度肾功能损害或需要透析的患者,在开始使用艾伏尼布片治疗之前,应考虑其风险和潜在获益。
老年人
老年患者无需进行剂量调整(参见【老年用药】)。
儿童
尚无本品用于 18 岁以下患者的临床研究资料。
不良反应:
下列临床重要的不良反应参见【注意事项】
● 分化综合征
● QTc 间期延长
● 格林巴利综合征
临床试验经验
由于临床试验是在各种不同的条件下进行,所以在药物临床试验中观察到的不良反应发生率不能直接与另一种药物临床试验的不良反应的发生率进行比较, 并且可能无法反映其实际应用中的发生率。
在研究 AG120-C-001 中,对 213 例 AML 患者每日 500 mg 艾伏尼布片单药治疗的安全性进行了评价。接受艾伏尼布片治疗的患者的中位年龄为 68 岁(年龄范围 18-87),其中 68% 的患者为 65 岁或以上,男性占 51%,白人占 66%,黑人或非裔美国人占 6%,亚洲人占 3%,夏威夷土著或其他太平洋岛民占 0.5%, 美洲印第安人或阿拉斯加原住民占 0.5%,其他未提供占 24%。在接受艾伏尼布 片治疗的 213 例患者中,37% 的患者暴露持续时间至少 6 个月,14% 的患者暴露 持续时间至少 1 年。在 213 例接受艾伏尼布片治疗的患者中,最常见的包含实验 室异常的不良反应( ≥ 20%)为血红蛋白降低、疲乏、关节痛、血钙降低、血钠 降低、白细胞增多症、腹泻、血镁降低、水肿、恶心、呼吸困难、尿酸升高、血钾降低、碱性磷酸酶升高、粘膜炎、天门冬氨酸氨基转移酶升高、血磷酸盐降低、 心电图 QT 间期延长、皮疹、血肌酐升高、咳嗽、食欲下降、肌痛、便秘和发热。
复发性或难治性 AML对 AG120-C-001 中接受艾伏尼布片单药每日 500 mg 治疗的 179 例复发性或难治性 AML 成人患者的安全性特征进行了研究。
本品的中位暴露持续时间为 3.9 个月(范围 0.1-39.5 个月)。65 例患者(36%) 的暴露持续时间至少 6 个月,16 例患者(9%)的暴露持续时间至少 1 年。
严重不良反应( ≥ 5%)为分化综合征(10%)、白细胞增多症(10%)和心电 图 QT 间期延长(7%)。有 1 例患者发生进行性多灶性白质脑病(PML)。
最常见的导致暂停给药的不良反应为心电图 QT 间期延长(7%)、分化综合征(3%)、白细胞增多症(3%)和呼吸困难(3%)。179 例患者中,5 例患者(3%) 因不良反应降低剂量。导致剂量降低的不良反应包括心电图 QT 间期延长(1%)、 腹泻(1%)、恶心(1%)、血红蛋白降低(1%)和转氨酶升高(1%)。导致永久停药的不良反应包括格林巴利综合征(1%)、皮疹(1%)、口腔粘膜炎(1%)和 肌酐升高(1%)。
临床试验中报告的最常见的不良反应如表 2 所示。
表 2. ≥ 10%(任何级别)或 ≥ 5%( ≥ 3 级)复发性或难治性 AML 患者报告的不良反应
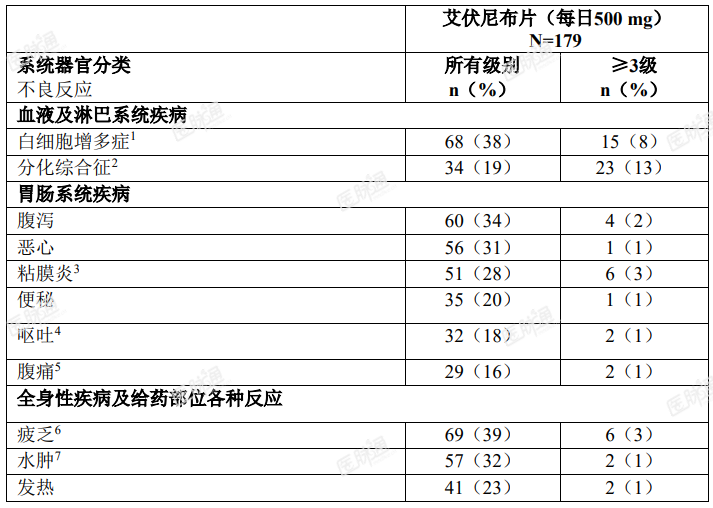
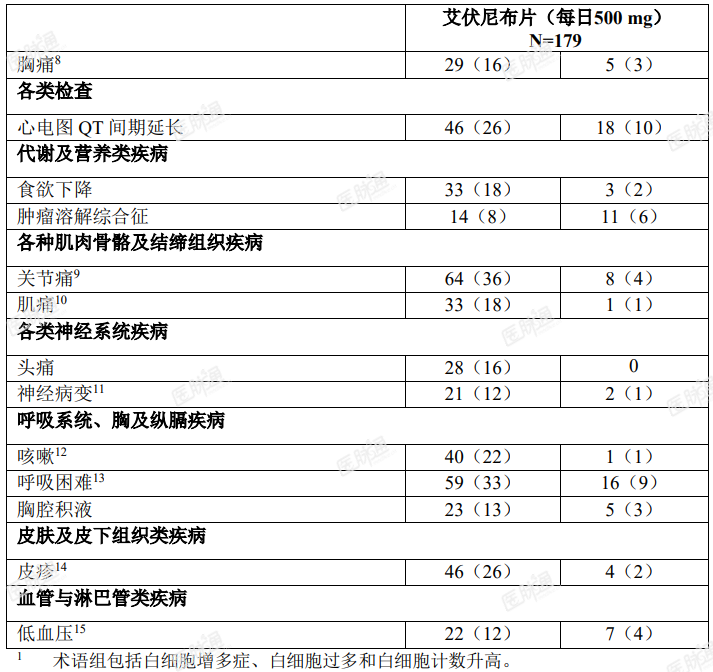


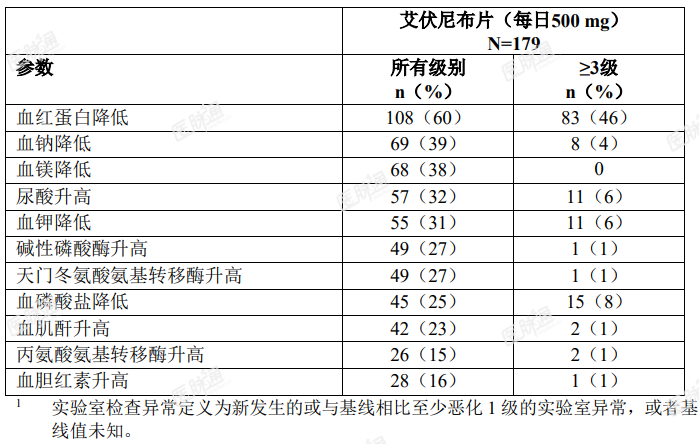
在复发性或难治性 AML 患者中观察到的特定基线后实验室数值变化如表 3 所示。
表 3. 复发性或难治性 AML 患者中报告的最常见( ≥ 10%)或 ≥ 5%( ≥ 3 级)新发生的或恶化的实验室检查异常 1
禁忌:
对本品活性成份或任何辅料过敏者禁用。
注意事项:
分化综合征
在临床试验中,19%(34/179)的复发性或难治性 AML 患者接受艾伏尼布片治疗后发生分化综合征。
分化综合征与骨髓细胞快速增殖和分化有关,如果不进行治疗可能危及生命或导致死亡。本品治疗的患者中分化综合征症状包括非感染性白细胞增多症、外周水肿、发热、呼吸困难、胸腔积液、低血压、缺氧、肺水肿、肺部炎症、心包积液、皮疹、体液过多、肿瘤溶解综合征和肌酐升高。34 例发生分化综合征的复发性或难治性 AML 患者中,27 例(79%)在治疗或暂停本品给药后恢复。分化综合征在开始本品治疗的第 1 天至 3 个月内发生,不一定伴有白细胞增多症。
如果怀疑发生分化综合征,给予地塞米松 10 mg,每 12 小时一次静脉输注(或等效剂量的口服或静脉输注皮质类固醇替代治疗),并进行血液动力学监测,直至缓解(参见【用法用量】)。如果伴发非感染性白细胞增多症,则根据临床指征给予羟基脲治疗或者进行白细胞去除术。在症状恢复并给予至少 3 天皮质类固醇治疗后,可逐渐减少皮质类固醇和羟基脲治疗。过早终止皮质类固醇和或羟基脲的治疗可能会导致分化综合征症状反复。如果给予皮质类固醇治疗后相关重度症状和或体征持续超过 48 小时级别不改善,则暂停本品治疗,直至相关重度症状和体征改善至非重度(参见【用法用量】)。
QTc 间期延长
本品治疗的患者可能出现 QT(QTc)间期延长和室性心律失常。在临床试验 258 例接受本品治疗的恶性血液疾病患者中,9% 的患者的 QTc 间期大于 500 msec,14% 的患者与基线 QTc 相比增加 60 msec。1 例患者发生了与本品相关的心室颤动。该项临床试验排除了基线 QTc ≥ 450 msec(除非 QTc ≥ 450 msec 是由预先存在的束支传导阻滞引起)或有长 QT 综合征或未控制的或严重心血管疾病病史的患者。
本品与已知可延长 QTc 间期的药物(如,抗心律失常药、氟喹诺酮类、三唑类抗真菌药、5-HT3 受体拮抗剂)和 CYP3A4 抑制剂合并使用可能增加 QTc 间期延长的风险(参见【药物相互作用】)。注意监测心电图(ECG)和电解质(参见【用法用量】)。
患有先天性长 QTc 综合征、充血性心力衰竭、电解质异常或正在服用已知可延长 QTc 间期药物的患者,可能需要增加监测频率。
如果 QTc 增加至大于 480 msec 且小于 500 msec,应暂停本品治疗。如果 QTc 增加至大于 500 msec,应暂停本品治疗并后续减量。对发生 QTc 间期延长并伴发危及生命的心律失常体征或症状的患者,应永久停用本品(参见【用法用量】)。
格林巴利综合征
在临床研究中,<1%(2/258)的接受本品治疗的患者发生格林巴利综合征。
应监测服用本品的患者是否出现运动和或感觉神经病变的新体征或症状,比如单侧或双侧无力、感觉改变、感觉异常或呼吸困难。确诊患有格林巴利综合征的患者,应永久停用本品(参见【用法用量】)。
低钾血症
在中国临床研究中,50%(15/30)的接受本品治疗的患者发生低钾血症。在治疗前及治疗期间应常规监测患者的电解质水平,并注意是否存在低钾血症。对于治疗前及治疗过程中发生低钾血症的患者,应予以及时补充纠正,并加强电解质的监测。对于存在低钾血症的患者,建议同时关注患者是否存在 QTc 间期延长。并根据与治疗相关的其他毒性的总体指导原则及 QTc 间期延长的指导原则,对本品进行必要的减量及暂停。(参见【用法用量】)。
孕妇及哺乳期妇女用药:
避孕
具有生育能力的女性患者、具有生育能力女性伴侣的男性患者应在治疗期间及末次给药后至少 1 个月内使用有效避孕方法。与本品合并给药可能降低激素避孕药的浓度,接受本品治疗的患者应在治疗期间及末次给药后至少 1 个月内使用其他的避孕方法。 具有生育能力的女性应在开始本品治疗之前接受妊娠检测。
孕妇
妊娠期女性接受本品治疗可能对胎儿造成伤害。如果在妊娠期间服用本品,或者患者在服药期间怀孕,应告知患者其对胎儿的潜在风险。
哺乳期
暂无人乳中本品或代谢物、对母乳喂养幼儿的影响或者对乳汁产量的影响的数据。由于许多药物可分泌到人乳中,并且母乳喂养幼儿可能发生不良反应,所以,建议在本品治疗期间和最后一次给药后至少 1 个月停止哺乳。
生育力
本品尚未进行动物和人体生育力毒性研究。
儿童用药:
尚未确定本品在 18 岁以下患者中的安全性和有效性。
老年用药:
在临床研究中,179 例复发性或难治性 AML 患者中有 112 例(63%)年龄在 65 岁或以上,有 40 例(22%)患者在 75 岁或以上。在 65 岁及以上复发性或难治性 AML 患者与年轻患者之间未观察到有效性或安全性方面的总体差异。
药物相互作用:
其他药物对艾伏尼布片的影响
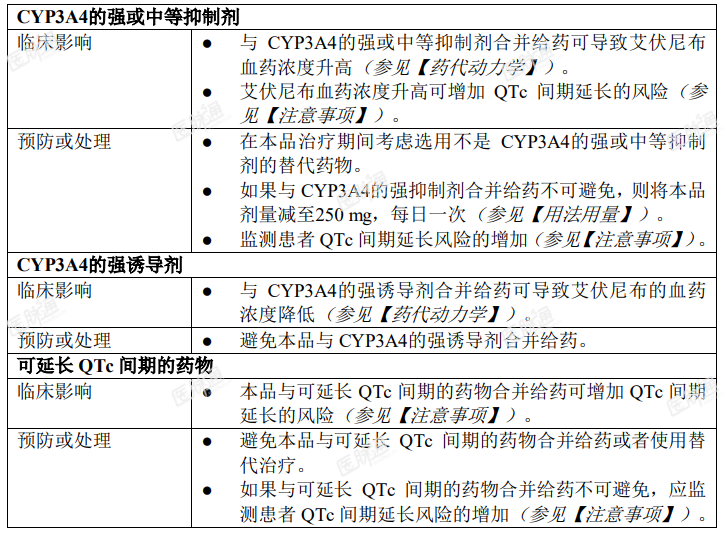
艾伏尼布片对其他药物的影响
本品对 CYP3A4 有诱导作用,可能对 CYP2C9 也有诱导作用。合并给药将降低属于 CYP3A4 敏感底物的药物的浓度,并可能降低属于 CYP2C9 敏感底物的药物浓度(参见【药代动力学】)。因此,在本品治疗期间建议改用非 CYP3A4 和 CYP2C9 敏感底物的药物。预期本品可降低抗真菌药物疗效,故本品不能与伊曲康唑或酮康唑(CYP3A4 底物)同时使用。与本品合并给药可能降低激素避孕药的浓度,接受本品治疗的患者应考虑其他避孕方法。如果本品与 CYP3A4 或 CYP2C9 敏感底物合并给药不可避免,应监测这类药物的疗效有否降低。
药物过量:
尚未开展相关研究。
临床试验:
本品的疗效评估是基于一项开放性、单臂、多中心临床试验(研究 AG120-C001,NCT02074839)中 174 例携带 IDH1 突变的成人复发性或难治性 AML 患者的数据。采用当地或中心检测确定 IDH1 突变,并使用 Abbott RealTimeTM IDH1 检测进行回顾性确认,该检测经 FDA 批准用于选择 AML 患者接受本品治疗。本品是以每日 500 mg 的起始剂量口服给药,直到疾病进展、出现不可接受的毒性或进行造血干细胞移植。21/174 例(12%)患者在本品治疗后接受干细胞移植。
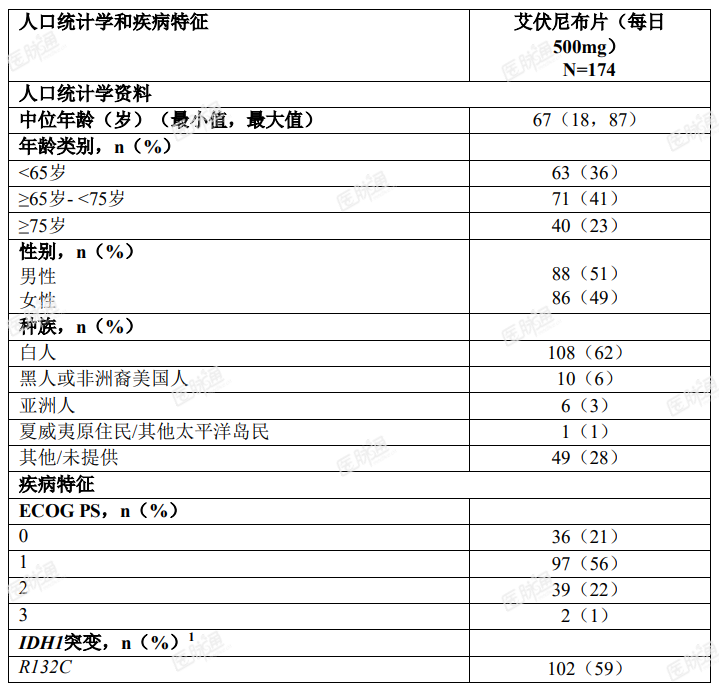
基线人口统计学和疾病特征如表 4 所示。
表 4. 复发性或难治性 AML 患者人口统计学和疾病基线特征
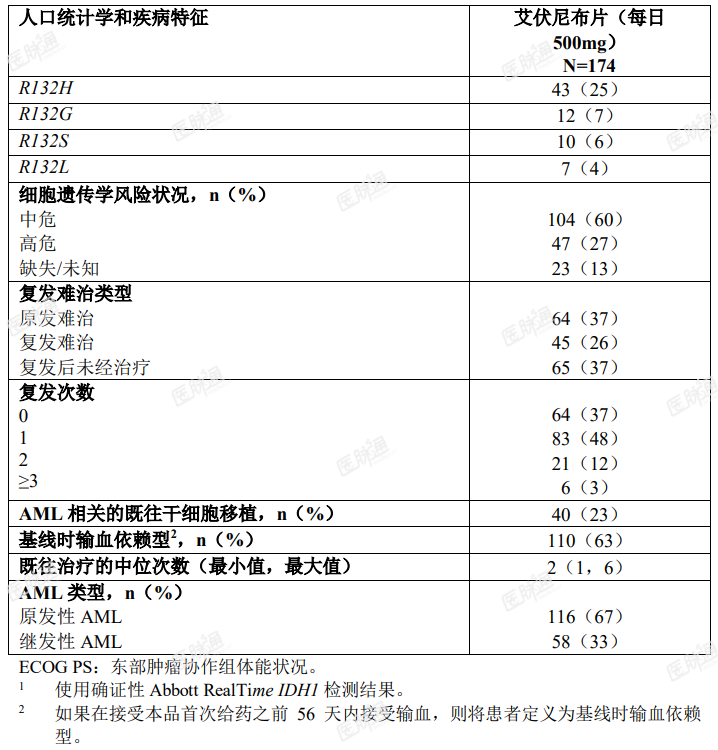
疗效的确定基于完全缓解(CR) + 伴有部分血液学恢复的完全缓解(CRh)的比率、CR + CRh 持续时间和从输血依赖型转换成非输血依赖型的比率。疗效结果如表 5 所示。中位随访时间为 8.3 个月(范围,0.2-39.5 个月),中位治疗持续时间为 4.1 个月(范围,0.1-39.5 个月)。
表 5. 复发性或难治性 AML 患者的疗效结果
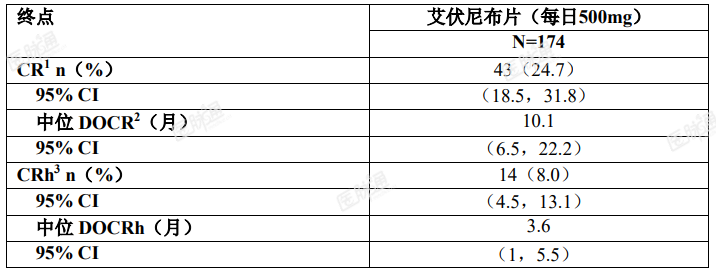
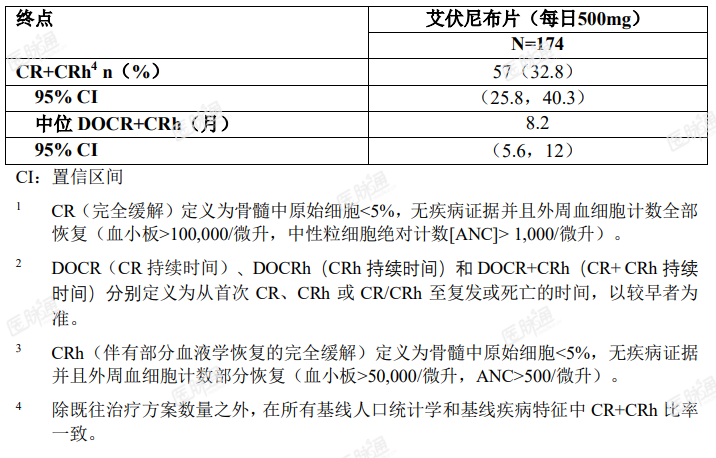
对达到 CR 或 CRh 的患者,其达到 CR 或 CRh 的中位时间为 2 个月(范围,0.9-5.6 个月)。在最佳缓解达到 CR 或 CRh 的 57 例患者中,所有患者均在开始本品治疗后的 6 个月内达到首次的 CR 或 CRh 应答。
在基线时红细胞(RBC)和或血小板输注依赖的 110 例患者中,41 例患者(37.3%)在基线后任意连续的 56 天转变并保持非输血依赖。在基线时 RBC 和血小板输注不依赖的 64 例患者中,38 例患者(59.4%)在基线后的任意连续 56 天期间保持非输血依赖。
本品基于境外一项单臂临床试验的缓解率以及中国受试者药代动力学数据获得附条件批准上市,治疗中国患者的有效性和安全性尚待上市后进一步确证。
贮藏:
密封,不超过 25 ℃ 保存。
包装:
高密度聚乙烯瓶,聚丙烯瓶盖包装,并加聚乙烯管装药用干燥剂。60 片瓶。
有效期:
48 个月
执行标准:
JX20220007
进口药品注册证号:
国药准字HJ20220002
委托方企业:
Servier Pharmaceuticals LLC生产企业:
PATHEON INC.
{kind=link}