031186508999
Xyb@xuekang.com

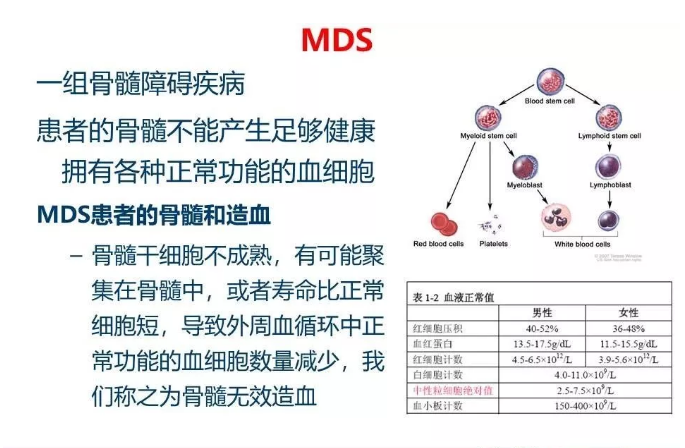
骨髓增生异常综合征(MDS)是一组异质性克隆性造血干细胞疾病,其临床特征是骨髓功能衰竭导致血细胞减少及其他相关并发症发生。在过去的几年里,从潜质未定的克隆性造血(CHIP)、意义不明的克隆性血细胞减少症(CCUS)到MDS,研究者们对克隆性造血障碍疾病有了更多的了解。
MDS的精确诊断需要从临床、病理和分子学等多方面进行综合分析,对于异常形态的病理表现,需要有经验的血液病理专家进行判断。下一步是疾病风险评估,根据不同疾病风险相应地调整治疗方案及治疗目标。修订的国际预后评分系统(R-IPSS)(根据血细胞减少及其程度、原始粒细胞百分比和细胞遗传学)是评估疾病风险最常用的工具,R-IPSS可以通过体细胞突变情况、患者的合并症和其他因素进行综合评估,超过一半的MDS患者最终被归为低危MDS(LR-MDS)。既往LR-MDS的治疗目标是减轻血细胞减少症以预防并发症,提高患者的生活质量,尽管治疗血细胞减少可能间接影响生存期,但迄今为止,没有治疗LR-MDS改善总生存期的明确证据。
在大多数LR-MDS中,治疗目标是改善贫血,红细胞生成刺激剂(ESA)是一种控制贫血较好的治疗选择,此外,可以根据红细胞(RBC)输血负荷和内源性血清促红细胞生成素预测ESA的治疗反应,并且需要足够的ESA治疗剂量和持续时间来评估反应。
对于del(5q)MDS患者,来那度胺是首选的治疗方法,具有较高的RBC输血独立性(TI)发生率。治疗早期的血细胞减少可预测且可控制。最近一项数据表明,较早和持续一定时间使用低剂量的来那度胺与较长的输血依赖时间和较高的细胞遗传学反应率相关。需要注意的是,del(5q)患者来那度胺治疗失败后的预后很差。
对于伴环状铁粒幼细胞(RS)亚型的LR-MDS患者,Luspatercept被批准用于治疗贫血,这是MDS十年来的首个获批的药物。Luspatercept是一种首创的红细胞成熟剂(EMA),可调节晚期红细胞的成熟。Luspatercept具有较高的RBC-TI发生率,特别是对于输血负担低的患者。相关研究正在探索这种新型药物(与ESA相比或与其联合)在LR-MDS患者的早期治疗中的疗效。
对于年轻的非del(5q)和非RS LR-MDS患者,在病程早期,骨髓细胞不足,抗胸腺细胞球蛋白(ATG)和环孢素的免疫抑制治疗(IST)可产生持久反应。对于那些不宜进行IST治疗的患者,如果孤立性贫血且血小板和中性粒细胞计数充足,那么来那度胺也可以作为治疗选择。
去甲基化剂药物(HMA)(阿扎胞苷和地西他滨)应该用于具有较高风险疾病特征、晚期病程或双/全血细胞减少症的患者,尤其是不适合进行IST治疗的患者。更短的治疗“3天方案”正在探索中,这可能是LR-MDS患者的一种新的治疗选择,口服HMA治疗LR-MDS的研究也正在进行中。此外,几种治疗LR-MDS的新型药物正在进行临床试验,Metlestat是一种端粒酶抑制剂,在II期临床研究中表现出令人鼓舞的活性,目前正在III期随机临床试验中进行研究。


{kind=link}